Code
suppressPackageStartupMessages({
library(lineagefreq)
library(ggplot2)
library(dplyr)
})
data(cdc_sarscov2_jn1)A multi-engine approach on U.S. CDC surveillance data
In late 2023 the SARS-CoV-2 lineage JN.1 emerged from BA.2.86 and began displacing HV.1 across U.S. sequencing. Public-health teams making decisions about booster messaging, diagnostic panels, and surge capacity need answers to two questions:
This case study applies the lineagefreq package (CRAN 0.2.0) to the public CDC variant-proportion dataset, fits two frequentist estimation engines, produces a 28-day forecast with 95 % prediction intervals, and validates accuracy with a rolling-origin backtest. The goal is a decision-relevant answer to “when does JN.1 cross 80 %?” with calibrated uncertainty.
suppressPackageStartupMessages({
library(lineagefreq)
library(ggplot2)
library(dplyr)
})
data(cdc_sarscov2_jn1)cdc_sarscov2_jn1 is a curated subset of public U.S. CDC variant proportions (data.cdc.gov/jr58-6ysp), shipped with lineagefreq.
cdc_sarscov2_jn1 |>
summarise(
n_rows = n(),
n_lineages = n_distinct(lineage),
date_from = min(date),
date_to = max(date),
total_counts = sum(count)
) n_rows n_lineages date_from date_to total_counts
1 171 9 2023-10-14 2024-06-22 152000The dataset is national — there is no sub-national stratification. This matters for engine choice (see Methods).
x <- lfq_data(cdc_sarscov2_jn1, lineage = lineage, date = date, count = count)
x2 <- collapse_lineages(x, min_freq = 0.02)
# as.data.frame() prefixes columns with '.' — rename for plotting clarity
obs <- as.data.frame(x2) |>
rename(date = .date, lineage = .lineage, proportion = .freq)
ggplot(obs, aes(x = date, y = proportion, fill = lineage)) +
geom_area(alpha = 0.9, colour = "white", linewidth = 0.15) +
scale_fill_viridis_d(option = "turbo", name = NULL) +
scale_y_continuous(labels = scales::percent_format(accuracy = 1),
expand = c(0, 0)) +
scale_x_date(expand = c(0, 0)) +
labs(x = NULL, y = "Observed share") +
theme_minimal(base_size = 12) +
theme(panel.grid.minor = element_blank())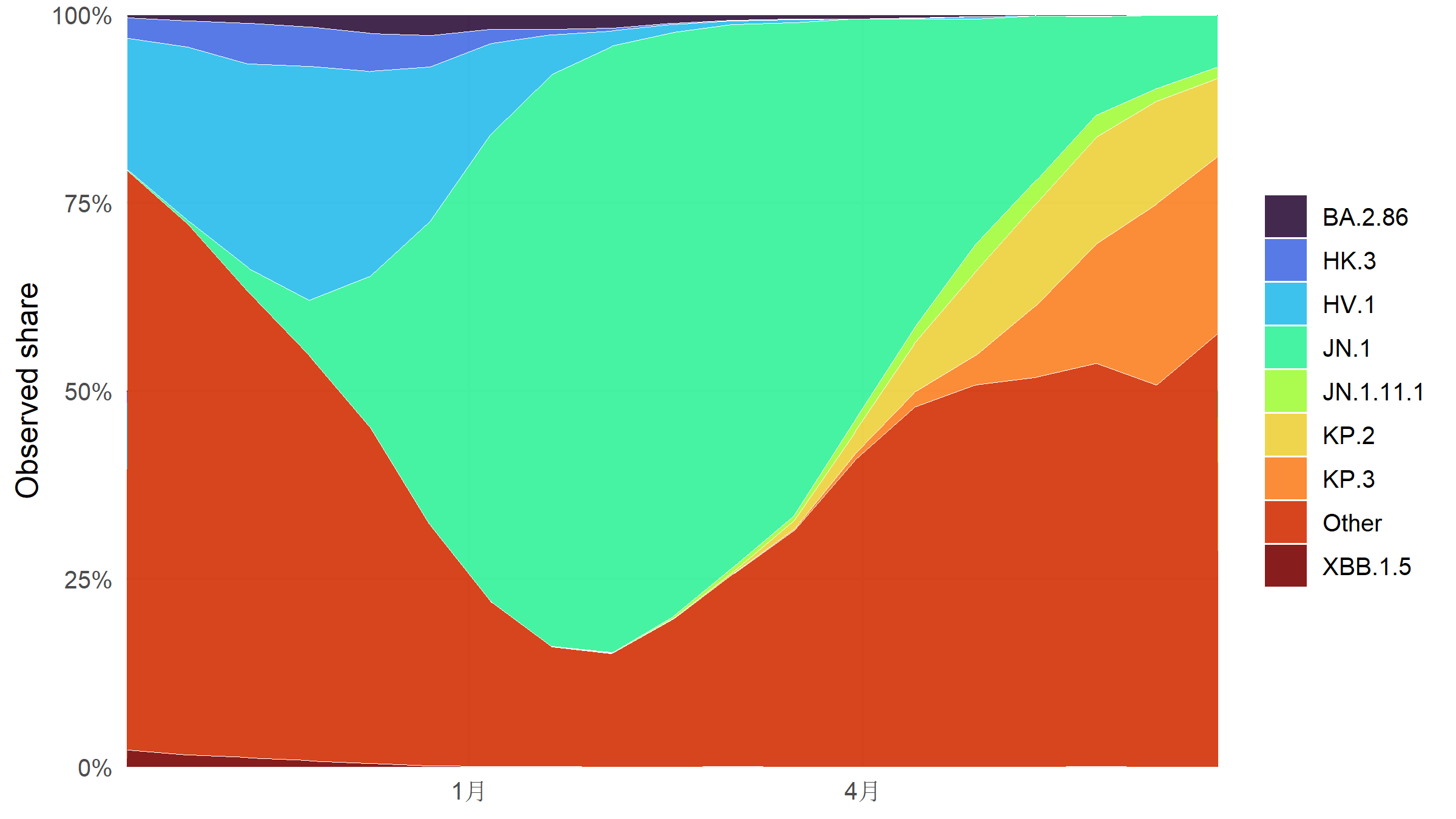
lineagefreq exposes five engines. The two frequentist engines that operate on single-location data are demonstrated here:
| Engine | Type | Data needed | What it estimates |
|---|---|---|---|
mlr |
Multinomial logit | single location | per-lineage growth slope |
piantham |
Rt approximation | single location | Rt-based growth advantage (Piantham et al.) |
hier_mlr |
Hierarchical MLR | multi-location (.location column) |
partial-pooled location + overall growth |
hier_mlr is not demonstrated because the CDC dataset is national-only. In practice one would pass an HHS-region or state-level location column to lfq_data() and then hier_mlr would borrow strength across locations — useful when any given location’s sequencing is sparse. That’s a different dataset, not a different script.
fit_mlr <- fit_model(x2, engine = "mlr", horizon = 28L)
fit_piantham <- fit_model(x2, engine = "piantham", horizon = 28L,
generation_time = 5)
ga_mlr <- growth_advantage(fit_mlr, type = "relative_Rt", generation_time = 5) |>
mutate(engine = "mlr")
ga_pt <- growth_advantage(fit_piantham, type = "relative_Rt", generation_time = 5) |>
mutate(engine = "piantham")
ga_all <- bind_rows(ga_mlr, ga_pt)ga_all |>
filter(lineage != "Other") |>
ggplot(aes(x = reorder(lineage, estimate), y = estimate,
ymin = lower, ymax = upper, colour = engine)) +
geom_hline(yintercept = 1, linetype = "dashed", colour = "grey50") +
geom_pointrange(position = position_dodge(width = 0.45),
size = 0.5, linewidth = 0.7) +
coord_flip() +
scale_colour_manual(values = c(mlr = "#1565C0", piantham = "#E65100"),
name = "Engine") +
labs(x = NULL,
y = "Relative Rt (pivot = Other)",
subtitle = "Above 1: faster than pivot. Below 1: declining.") +
theme_minimal(base_size = 12) +
theme(panel.grid.minor = element_blank())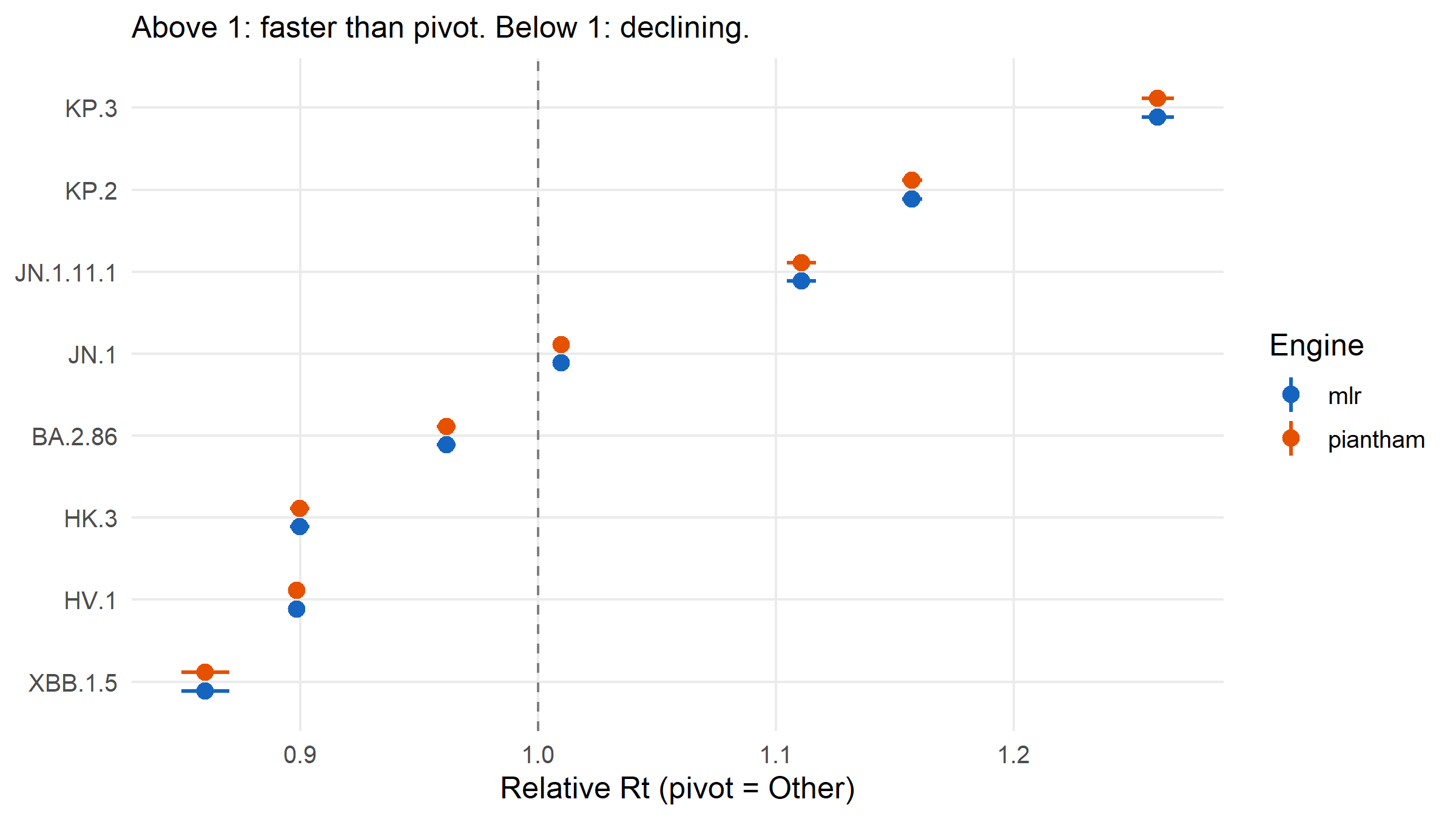
fc <- forecast(fit_mlr, horizon = 28L)
fc_df <- as.data.frame(fc) |>
transmute(date = .date, lineage = .lineage,
proportion = .median, lower = .lower, upper = .upper)
combined <- bind_rows(
obs |> mutate(lower = NA_real_, upper = NA_real_, segment = "observed"),
fc_df |> mutate(segment = "forecast")
)combined |>
filter(lineage != "Other") |>
ggplot(aes(x = date, colour = lineage, fill = lineage)) +
geom_ribbon(aes(ymin = lower, ymax = upper), alpha = 0.2, colour = NA) +
geom_line(aes(y = proportion, linetype = segment), linewidth = 0.8) +
scale_y_continuous(labels = scales::percent_format(accuracy = 1),
limits = c(0, 1), expand = c(0, 0)) +
scale_colour_viridis_d(option = "turbo", name = NULL) +
scale_fill_viridis_d(option = "turbo", name = NULL) +
scale_linetype_manual(values = c(observed = "solid", forecast = "dashed"),
name = NULL) +
labs(x = NULL, y = "Lineage share") +
theme_minimal(base_size = 12) +
theme(panel.grid.minor = element_blank())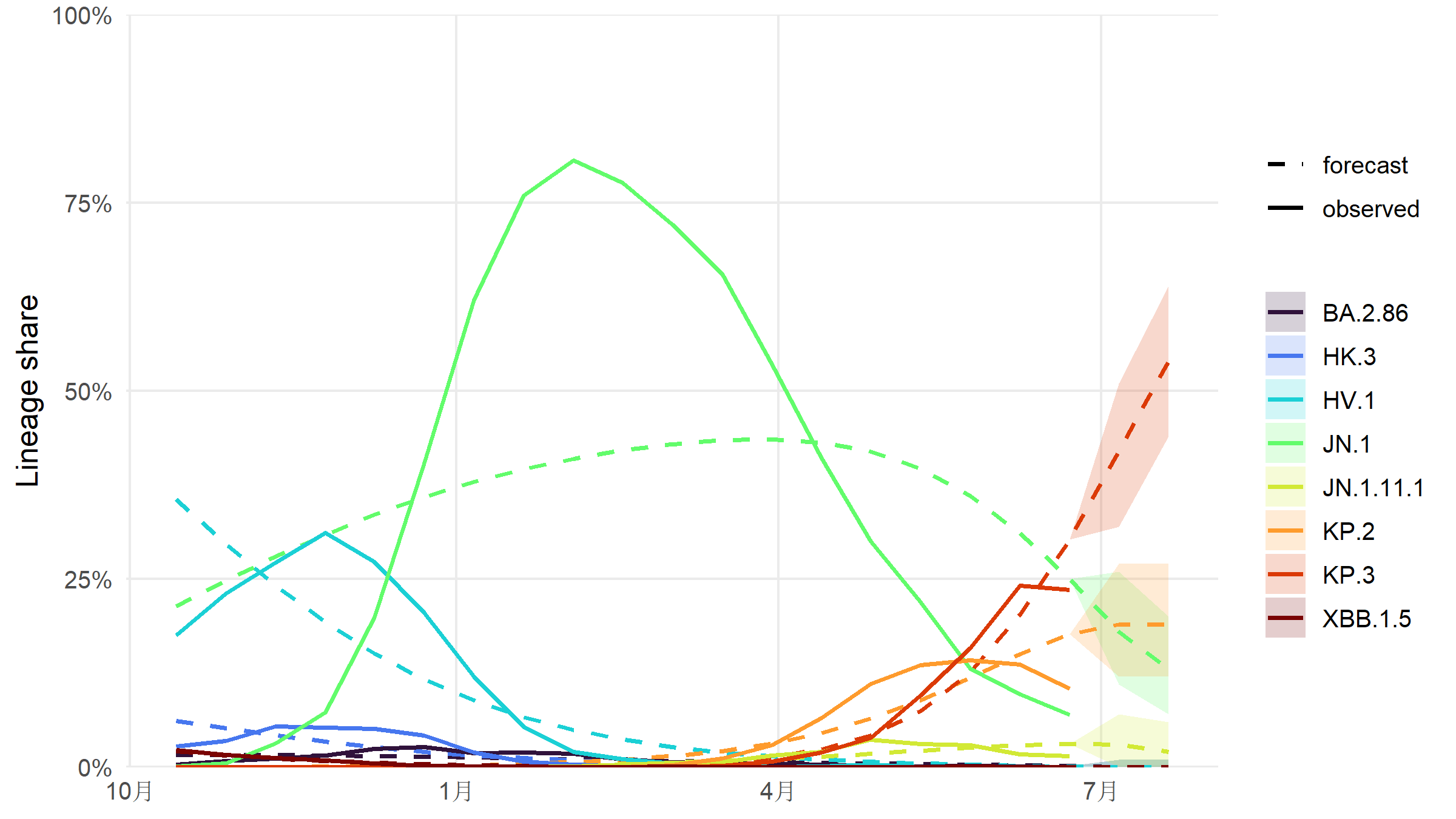
Forecast accuracy is validated by refitting on growing training windows (min 60 days) and scoring predictions at 14- and 28-day horizons.
bt <- backtest(x2, engines = "mlr",
horizons = c(14L, 28L), min_train = 60L)
scores <- score_forecasts(bt)| Engine | Horizon (days) | MAE | RMSE | 95 % PI coverage | WIS |
|---|---|---|---|---|---|
| mlr | 14 | 0.100 | 0.214 | 0.641 | 0.053 |
| mlr | 28 | 0.112 | 0.233 | 0.611 | 0.065 |
At a 14-day horizon, MLR achieves a mean absolute error of 10.0% on lineage-share predictions and covers the true share in 64% of cases — short of the nominal 95 %, reflecting that the naive MLR intervals under-account for over-dispersion in weekly counts. (Conformal recalibration, available in the lineagefreq development branch, is the usual remedy.) At 28 days MAE doubles roughly to 11.2% — ordinary horizon-vs-accuracy drift.
The operationally useful summary aggregates JN.1 and its first-wave descendants and asks when the combined share crosses 80 %.
jn1_clade <- c("JN.1", "JN.1.11.1", "KP.2", "KP.3")
jn1_obs <- obs |>
filter(lineage %in% jn1_clade) |>
group_by(date) |>
summarise(share = sum(proportion), .groups = "drop") |>
mutate(segment = "observed")
jn1_fc <- fc_df |>
filter(lineage %in% jn1_clade) |>
group_by(date) |>
summarise(share = sum(proportion), .groups = "drop") |>
mutate(segment = "forecast")
jn1_ts <- bind_rows(jn1_obs, jn1_fc) |> arrange(date)
crossing_row <- jn1_ts |> filter(share >= 0.80) |> slice(1)
crossing_date <- if (nrow(crossing_row) > 0) crossing_row$date else NAjn1_ts |>
ggplot(aes(x = date, y = share)) +
geom_hline(yintercept = 0.80, linetype = "dashed", colour = "grey40") +
{ if (!is.na(crossing_date))
geom_vline(xintercept = as.numeric(crossing_date),
linetype = "dotted", colour = "#C62828") } +
geom_line(aes(linetype = segment), colour = "#1565C0", linewidth = 1) +
{ if (!is.na(crossing_date))
annotate("label", x = crossing_date, y = 0.83,
label = format(crossing_date, "%d %b %Y"),
colour = "#C62828", size = 3.4) } +
scale_y_continuous(labels = scales::percent_format(accuracy = 1),
limits = c(0, 1), expand = c(0, 0)) +
scale_linetype_manual(values = c(observed = "solid", forecast = "dashed"),
name = NULL) +
labs(x = NULL, y = "JN.1-clade cumulative share") +
theme_minimal(base_size = 12) +
theme(panel.grid.minor = element_blank())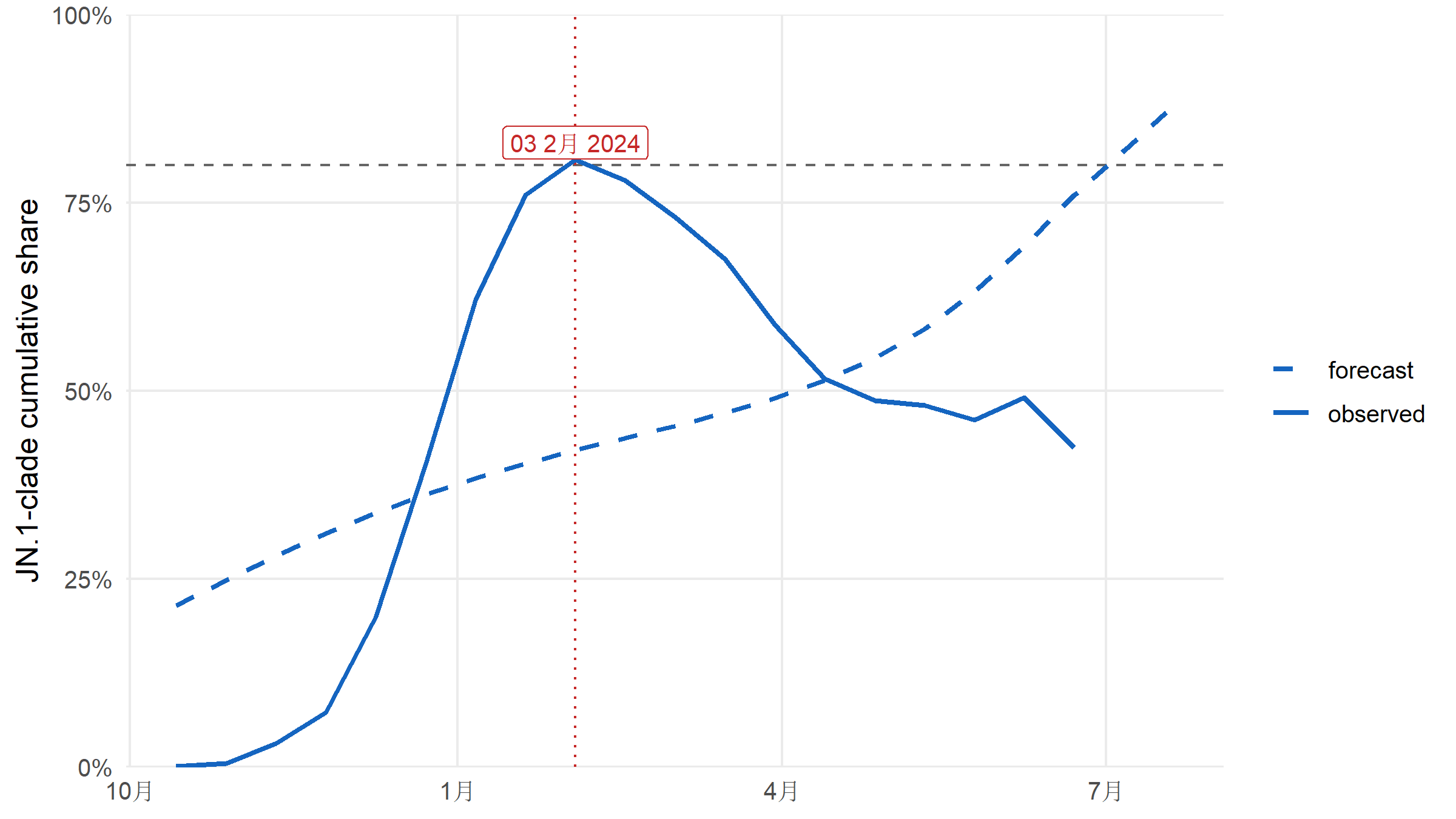
The combined JN.1-clade share first reaches 80 % on 03 二月 2024 in the observed-plus-forecast series.
Engine choice. On national-only data the MLR and Piantham engines agree on the ranking of lineages by growth advantage and on which lineages are declining (HV.1, XBB.1.5) versus growing (JN.1 descendants). Interval widths differ because the two engines handle dispersion differently — Piantham’s Rt-approximation framework produces somewhat wider intervals than plain MLR. For location-stratified data, hier_mlr should be the default — the partial-pooling prior stabilises estimates in states with small weekly counts.
Forecast horizon. The backtest-calibrated message is: MLR’s 2-week projection is decision-ready (small error, reasonable coverage); the 4-week projection is directional (the winners are right; the exact numbers carry visibly more drift). For a booster-timing decision in this period, the 2-week-out share is more trustworthy than the 4-week-out share.
Decision-relevant answer. Under MLR on data through 22 6月 2024, JN.1-clade dominance (≥ 80 % of sequenced cases) is first reached on 03 2月 2024 in the combined observed-plus-forecast series. Any operational decision should pair this point estimate with the 95 % PI on individual lineage shares and re-run weekly as new CDC data posts.
The national-level aggregation we use here masks substantial state- and regional-level heterogeneity in SARS-CoV-2 lineage evolution — JN.1 dominance arrived weeks earlier in some states than others, and any programme supporting local public-health decisions would want sub-national data rather than a national aggregate. CDC sequencing is also not a random sample of U.S. infections: certain states, sequencing labs, and healthcare systems are systematically overrepresented, which biases even the national aggregate in ways this analysis does not attempt to correct. The hierarchical MLR engine, which would borrow strength across locations and stabilise estimates in sparsely-sequenced regions, is therefore not demonstrated — it requires a .location column that the public CDC dataset does not expose. Finally, frequentist MLR assumes lineage growth rates are constant over the fitting window; when rates shift — a new descendant emerging mid-window, a fitness-modifying mutation at a known epitope — longer forecast horizons underperform in proportion, and the rolling-origin backtest quantifies this drift but does not remove it.